High-Dose Fenbendazole Protocol: Dosing, Safety, and Evidence
- Goal: Escalate fenbendazole dosing systematically while managing hepatotoxicity risk
- Duration: Monthly escalation cycles with ongoing liver monitoring
- Key compounds: Fenbendazole (FBZ), with fatty-meal co-administration for absorption
- Dose range: 222 mg/day (standard) → 444 mg/day (intermediate) → 1,000 mg/day (high-dose maximum)
- Cycles: 3 days on / 4 days off per week; escalate only if liver enzymes remain normal
- Monitoring: Baseline ALT/AST/bilirubin required; repeat at 4 weeks, then monthly
Overview
Fenbendazole (FBZ) is a benzimidazole anthelmintic used for decades in veterinary medicine to treat intestinal parasites. In recent years, it has attracted interest as a potential repurposed oncology agent following anecdotal case reports and a growing body of preclinical research. The standard self-administration protocol — popularized online and drawn from veterinary dosing conventions — uses 222 mg/day for three consecutive days each week. However, some individuals and emerging case literature have escalated to 444 mg or 1,000 mg/day, motivated by the drug’s poor oral bioavailability and the hypothesis that higher systemic exposure may be necessary to reach concentrations that demonstrate anticancer activity in cell-culture studies.
This article reviews the pharmacokinetic basis for dose escalation, the available evidence on safety and efficacy across doses, documented hepatotoxicity cases, and the liver monitoring protocol considered prudent for any regimen above standard dosing. The evidence base is primarily preclinical and anecdotal; fenbendazole has not been evaluated in prospective human clinical trials for any oncologic indication.
Fenbendazole is not approved by the FDA or EMA for human use in any indication. All dosing discussed here reflects self-administration patterns reported in the literature, not clinical recommendations. Consult a qualified healthcare professional before considering any such protocol.
Pharmacokinetic Basis for Dose Escalation
Fenbendazole has extremely poor water solubility, approximately 0.3 µg/mL under aqueous conditions, resulting in variable and generally low oral bioavailability after standard dosing. The drug is metabolized primarily via CYP2C19 and CYP2J2 in human liver microsomes, as established by Lee et al. (2013), and a portion undergoes oxidation to the bioactive metabolite oxfendazole, which retains anthelmintic and potentially anticancer properties. Interindividual pharmacokinetic variability is substantial, meaning that a given dose may produce negligible systemic exposure in one person and meaningful exposure in another.
The food effect on fenbendazole absorption is clinically significant. Research by McKellar, Galbraith, and Baxter (1993) in dogs demonstrated that concurrent ingestion of food significantly increases FBZ bioavailability regardless of the fat content of the meal. This finding has informed the practical recommendation to take FBZ immediately with or mixed into a fatty meal — such as avocado, olive oil, or full-fat dairy — to maximize systemic exposure.
A more recent approach to bioavailability enhancement involves pharmaceutical formulation. Ding et al. (2024) prepared a fenbendazole:methyl-β-cyclodextrin inclusion complex that increased aqueous solubility approximately 60,000-fold (from 0.3 µg/mL to 20.21 mg/mL). In dogs receiving 5 mg/kg orally, the complex increased FBZ relative bioavailability by 138% and oxfendazole bioavailability by 149% compared to standard FBZ powder. These enhanced formulations are not commercially available as human cancer therapies, but the data help explain the empirical motivation for dose escalation: poor baseline absorption may mean that even doubling or tripling the dose results in only a moderate increase in effective systemic exposure.
A review by Cray and Altman (2022) documented that FBZ tissue concentrations in rodents fed FBZ-containing diet are approximately 10-fold lower than the concentrations shown to kill tumor cells in vitro (around 1 µM). This bioavailability gap may partially explain why some patients escalate doses well beyond standard veterinary-derived amounts.
Dose Escalation Strategy
The following escalation framework is drawn from doses referenced in published case reports and safety assessments. It is not a clinical protocol endorsed by any regulatory body. Escalation should only proceed if liver enzyme values remain within acceptable limits at each prior stage.
| Stage | Daily Dose | Schedule | Monitoring Action |
|---|---|---|---|
| Baseline (Weeks 1–2) | 222 mg | 3 on / 4 off | Baseline ALT, AST, bilirubin before starting |
| Intermediate (Weeks 3–4) | 444 mg | 3 on / 4 off | Repeat LFTs at 4 weeks; escalate only if normal |
| High-Dose (Month 2+) | 1,000 mg (maximum) | 3 on / 4 off | Monthly LFTs; hold if ALT/AST >3× ULN |
| Dose Reduction Threshold | Return to prior stage | — | ALT or AST >2× baseline or >3× ULN |
| Discontinuation Threshold | Stop entirely | — | ALT or AST >5× ULN, jaundice, or symptomatic hepatitis |
The 3-days-on / 4-days-off schedule is derived from veterinary antiparasitic dosing conventions and is referenced across the published case literature. The rationale for this cycling pattern as a cancer protocol is not clearly established from pharmacokinetic or pharmacodynamic data; it appears to have originated empirically and persisted through community adoption. Some patients reported in case series used continuous daily dosing, but this approach carries higher hepatotoxicity risk, particularly above 444 mg/day.
Mechanism of Action
Preclinical research suggests fenbendazole exerts anticancer effects through at least four distinct cellular mechanisms. The breadth of these pathways has been cited as a potential advantage over single-target agents, as simultaneous pressure on multiple cancer survival mechanisms may limit the ability of tumor cells to develop resistance through single-pathway escape.
Microtubule destabilization: FBZ binds the colchicine-binding site on β-tubulin and causes moderate microtubule depolymerization at micromolar concentrations. The landmark study by Dogra, Kumar, and Mukhopadhyay (2018) characterized this action as that of a “moderate destabilizer,” producing G2/M cell cycle arrest followed by mitotic slippage and apoptosis. This mechanism is distinct from highly potent agents such as colchicine or vinca alkaloids, and the apparent selectivity for cancer cells over normal cells may relate to altered tubulin isoform expression in tumor tissue.
Glucose metabolism inhibition: FBZ downregulates GLUT glucose transporters and hexokinase II (HKII) activity, reducing aerobic glycolysis — the metabolic reprogramming known as the Warburg effect that many cancers rely on for rapid energy production. In A549 lung cancer cells, FBZ inhibited glucose uptake as measured by 2-NBDG assay and reduced lactate production. This mechanism overlaps with and may be complementary to other metabolic-targeting agents.
p53 reactivation and stabilization: FBZ stabilizes p53 protein and promotes its nuclear accumulation, activating the p53-p21 pathway. Benzimidazoles also appear to downregulate Mdm2 and MdmX — the primary negative regulators of p53 — potentially restoring p53 activity in tumors where it has been functionally suppressed but not mutated. This effect has been demonstrated in melanoma cells overexpressing MdmX.
Proteasome impairment: Separate work by Dogra and Mukhopadhyay (2012) showed that fenbendazole inhibits the chymotrypsin-like, trypsin-like, and post-acidic activities of the 26S proteasome. Proteasome inhibition induces endoplasmic reticulum stress, reactive oxygen species (ROS) production, decreased mitochondrial membrane potential, and cytochrome c release, all of which contribute to apoptosis. This mechanism appears particularly relevant in small cell lung cancer cells.
Preclinical Efficacy Evidence
Dogra, Kumar, and Mukhopadhyay (2018) reported that 1 µM FBZ caused significant cancer cell death in A549 and H460 non-small cell lung cancer (NSCLC) lines in vitro, and that 1 mg/mouse administered orally every other day suppressed tumor growth and vascularity in xenograft models. The estimated human equivalent of the effective in vivo dose appears substantially lower than the doses empirically used in self-administration protocols, a discrepancy that likely reflects the poor oral bioavailability discussed above.
A broad review by Son, Lee, and Adunyah (2020) found that fenbendazole demonstrated activity across colorectal, lung, breast, prostate, lymphoma, and leukemia preclinical models, but also highlighted the inconsistency of in vivo translation. A murine lymphoma xenograft study found no in vivo antitumor effect despite promising in vitro results, illustrating the difficulty of extrapolating cell-culture findings to animal models — let alone humans.
Earlier work by Duan, Liu, and Rockwell (2013) in the EMT6 breast tumor model demonstrated that while FBZ was cytotoxic to cancer cells in vitro (with toxicity increasing under hypoxic conditions), maximally intensive in vivo regimens showed no significant antitumor benefit beyond additive cytotoxicity when combined with radiation or docetaxel. This study provided early evidence that the in vitro to in vivo translation for FBZ is inconsistent.
Comparison to Mebendazole Dosing in Clinical Trials
Mebendazole (MBZ), a structurally related benzimidazole, has been investigated in clinical trials for cancer, providing the most relevant human safety data for the compound class. Mansoori et al. (2021) conducted a Phase 2a clinical study in which 11 patients with advanced gastrointestinal cancer received individualized-dose MBZ at up to 4 g/day. The regimen was well tolerated, with no dose-limiting toxicities and no significant changes in liver enzyme values. However, all 10 treated patients had progressive disease, and the study was stopped early due to lack of efficacy.
This trial is relevant to high-dose FBZ protocols for two reasons. First, it establishes that benzimidazoles at doses up to 4 g/day are tolerable in short-term use under clinical supervision. Second, it underscores that tolerability does not translate to efficacy: even at doses four times higher than the highest commonly self-administered FBZ dose, the benzimidazole class did not produce objective responses in advanced GI cancer. The review by Nguyen et al. (2024) in Anticancer Research provides additional context on the broader clinical landscape of FBZ and related agents across human and veterinary oncology settings.
- Food effect: Co-administration with food significantly increases FBZ bioavailability regardless of fat content (McKellar et al. 1993)
- Cyclodextrin enhancement: Methyl-β-cyclodextrin complexation increased FBZ bioavailability by 138% in dogs (Ding et al. 2024)
- Bioavailability gap: Tissue FBZ concentrations in rodents are ~10× lower than in vitro anticancer concentrations, motivating dose escalation attempts (Cray & Altman 2022)
- MBZ clinical trial: Mebendazole up to 4 g/day was tolerable but showed no clinical efficacy in advanced GI cancer (Mansoori et al. 2021)
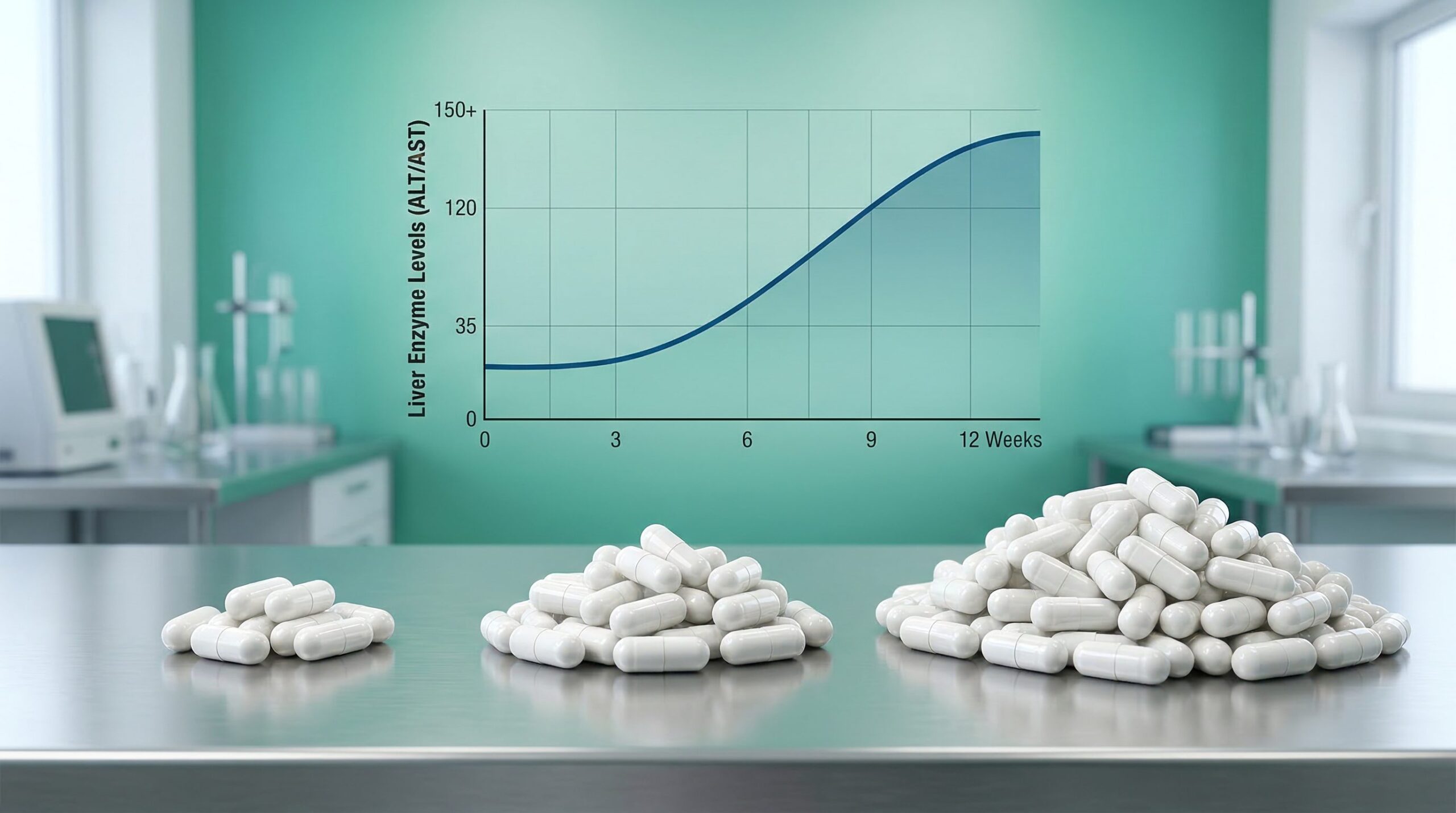
- DILI cases: At least two severe hepatotoxicity cases documented at ~1 g × 3/week; one required three months for liver enzyme normalization (Thakurdesai et al. 2024)
- EMA single-dose data: Single oral doses up to 2,000 mg and 500 mg/day for 10 days were well tolerated in incidental human exposure assessments (EMA, 2011)
Hepatotoxicity: Cases and Risk Factors
Drug-induced liver injury (DILI) from fenbendazole is not hypothetical — it has been documented in peer-reviewed case reports at doses commonly used in self-administration protocols. Both published severe DILI cases resolved after discontinuation of FBZ, but the recovery period was prolonged.
Thakurdesai et al. (2024) reported the first histologically confirmed severe DILI attributed to self-administered fenbendazole. A 67-year-old woman took 1 g sachets three times per week for approximately one year. She presented with jaundice and laboratory values showing AST of 1,869 U/L, ALT of 2,600 U/L, peak bilirubin of 24 mg/dL, and an INR of 1.6. Liver biopsy confirmed centrilobular hepatocyte necrosis with lymphocytic infiltrate. Treatment with N-acetylcysteine and budesonide was initiated; liver function tests normalized within approximately three months after FBZ discontinuation. This case establishes that the 1 g × 3/week dosing pattern carries a clear and serious hepatotoxic liability, with injury potentially developing over months of continuous use before clinical symptoms appear.
Yamaguchi et al. (2021) described severe hepatocellular DILI in an 80-year-old woman with advanced NSCLC who was concurrently receiving pembrolizumab (a checkpoint inhibitor immunotherapy) and self-administered fenbendazole at 1,000 mg/day, 3 days on / 4 days off, for approximately one month. AST peaked at 386 U/L and ALT at 487 U/L. Liver injury resolved after FBZ was stopped. Notably, no tumor shrinkage was observed. The concurrent use of pembrolizumab — which carries its own risk of immune-mediated hepatitis — complicated causal attribution, but the authors considered FBZ the primary contributing agent.
The mechanism of FBZ-associated hepatotoxicity is not fully established. CYP2C19 and CYP2J2-mediated metabolism generates reactive intermediates that may cause direct hepatocellular injury, as established by Lee et al. (2013). Risk factors include continuous high-dose use (above approximately 666 mg/day without breaks), concurrent immunotherapy, and pre-existing liver disease. Early warning signs — jaundice, dark urine, right upper quadrant pain, and fatigue — may be preceded by weeks of asymptomatic liver enzyme elevation, making regular blood testing the only reliable safety check.
An additional interaction concern was identified by Gardner et al. (2012), who demonstrated that fenbendazole dramatically potentiated acetaminophen (APAP) hepatotoxicity in mice. The clinical relevance of this finding in humans is uncertain, but it warrants caution in patients who use acetaminophen-containing products concurrently.
Important Considerations
The safety profile of high-dose fenbendazole is incompletely characterized. Human safety data derive primarily from the EMA’s assessment of Panacur AquaSol (EMEA/V/C/2008, December 2011), which documented that single oral doses up to 2,000 mg produced no adverse effects, and that 500 mg/day for 10 consecutive days was similarly tolerated in individuals incidentally exposed. These data come from acute or short-duration exposure assessments, not from chronic oncologic use.
Regarding drug interactions, FBZ is metabolized by CYP2C19 and CYP2J2; inhibitors of these enzymes — including omeprazole, fluconazole, and certain antidepressants — may increase FBZ plasma exposure. The potential for additive hepatotoxicity with concurrent checkpoint inhibitor immunotherapy is clinically relevant, given the case reported by Yamaguchi et al. Any patient receiving active cancer treatment should disclose FBZ use to their oncology team.
The evidence base for FBZ as a human anticancer agent rests primarily on preclinical data and a small number of case reports. A three-patient case series published by Makis, Baghli, and Martinez in 2025 — which described apparent remissions in patients self-administering fenbendazole alongside vitamins and other supplements — was retracted by the journal in January 2026. All positive anecdotal cases involve confounders, including concurrent conventional cancer therapy, multiple supplements, and lifestyle changes. No prospective clinical trial has evaluated FBZ dosing, safety, or efficacy for any cancer indication in humans.
This protocol has not been evaluated in formal clinical trials as a high-dose regimen. The information presented is for educational purposes only. Always consult a qualified healthcare professional before starting any new treatment protocol.
Documented hepatotoxicity has occurred at 1,000 mg × 3 days/week, including one case presenting after 12 months of use with peak ALT of 2,600 U/L. Liver enzyme monitoring is not optional at elevated doses. Patients with pre-existing liver disease, concurrent immunotherapy, or hepatic metastases face substantially higher risk and should not attempt dose escalation without direct hepatology supervision.
- Dogra N, Kumar A, Mukhopadhyay T. Fenbendazole acts as a moderate microtubule destabilizing agent and causes cancer cell death by modulating multiple cellular pathways. Sci Rep. 2018;8:11926. doi: 10.1038/s41598-018-30158-6. PMID: 30093705. PubMed
- Dogra N, Mukhopadhyay T. Impairment of the Ubiquitin-Proteasome Pathway by Methyl N-(6-Phenylsulfanyl-1H-benzimidazol-2-yl)carbamate Leads to a Potent Cytotoxic Effect in Tumor Cells. J Biol Chem. 2012;287(36):30625–30634. doi: 10.1074/jbc.M111.324228. PMID: 22787140. PubMed
- Lee DY, et al. CYP2J2 and CYP2C19 Are the Major Enzymes Responsible for Metabolism of Albendazole and Fenbendazole in Human Liver Microsomes and Recombinant P450 Assay Systems. Antimicrob Agents Chemother. 2013;57(12):5769–5777. doi: 10.1128/AAC.00843-13. PMID: 24041879. PubMed
- McKellar QA, Galbraith EA, Baxter P. Oral absorption and bioavailability of fenbendazole in the dog and the effect of concurrent ingestion of food. J Vet Pharmacol Ther. 1993;16(2):189–198. doi: 10.1111/j.1365-2885.1993.tb00163.x. PubMed
- Ding Y, Zhang Z, Ding C, Xu S, Xu Z. Preparation and evaluation of fenbendazole methyl-β-cyclodextrin inclusion complexes. BMC Vet Res. 2024;20:214. doi: 10.1186/s12917-024-04056-1. PMID: 38769544. PubMed
- Thakurdesai A, Rivera-Matos L, Nagra N, Busch B, Mais DD, Cave MC. Severe Drug-Induced Liver Injury Due to Self-administration of the Veterinary Anthelmintic Medication, Fenbendazole. ACG Case Rep J. 2024;11(5):e01354. doi: 10.14309/crj.0000000000001354. PMID: 38706451. PubMed
- Yamaguchi T, Shimizu J, Oya Y, Horio Y, Hida T. Drug-Induced Liver Injury in a Patient with Nonsmall Cell Lung Cancer after the Self-Administration of Fenbendazole Based on Social Media Information. Case Rep Oncol. 2021;14(2):886–891. doi: 10.1159/000516276. PMID: 34248555. PubMed
- Mansoori S, Fryknäs M, Alvfors C, Loskog A, Larsson R, Nygren P. A phase 2a clinical study on the safety and efficacy of individualized dosed mebendazole in patients with advanced gastrointestinal cancer. Sci Rep. 2021;11:8981. doi: 10.1038/s41598-021-88433-y. PMID: 33903692. PubMed
- Duan Q, Liu Y, Rockwell S. Fenbendazole as a potential anticancer drug. Anticancer Res. 2013;33(2):355–362. PMID: 23393327. PubMed
- Son D, Lee E, Adunyah SE. The Antitumor Potentials of Benzimidazole Anthelmintics as Repurposing Drugs. Immune Netw. 2020;20(4):e29. doi: 10.4110/in.2020.20.e29. PMID: 32895621. PubMed
- Cray C, Altman N. An Update on the Biologic Effects of Fenbendazole. Comp Med. 2022;72(4):229–238. doi: 10.30802/AALAS-CM-22-000006. PMID: 36039505. PubMed
- Gardner CR, Laskin JD, Laskin DL, Mishin V. Exacerbation of acetaminophen hepatotoxicity by the anthelmentic drug fenbendazole. Toxicol Sci. 2012;125(2):607–612. doi: 10.1093/toxsci/kfr301. PMID: 22100501. PubMed
- Nguyen J, Nguyen TQ, Han BO, Hoang BX. Oral Fenbendazole for Cancer Therapy in Humans and Animals. Anticancer Res. 2024;44(9):3725–3735. doi: 10.21873/anticanres.17197. PMID: 39197912. PubMed
- European Medicines Agency (EMA/CVMP). Panacur AquaSol EPAR — Public Assessment Report. EMEA/V/C/2008. December 2011. EMA